Contact Hours: 6
This online independent study activity is credited for 6 contact hours at completion.
Course Purpose
To provide healthcare professionals with knowledge of pharmacodynamics and pharmacokinetics of drugs when administered by various routes for various durations.
Overview
Drugs are used to prevent, cure, or control various diseases. To achieve this goal, adequate concentrations of the drug must be delivered to the target tissues so that therapeutic levels are obtained. Pharmacological and toxicological actions of drugs are mainly related to their plasma concentrations. Consequently, healthcare professionals who administer drugs must recognize the onset of a drug’s action as well as the intensity and duration of its effect. In turn, these are controlled by four fundamental pathways of drug movement and modification in the body, namely absorption, distribution, metabolism, and excretion. This learning content provides an overview of pharmacodynamics and pharmacokinetics through the identification of common definitions and terminology and provides a brief overview of the effects of a drug in single versus two compartment model administration.
Objectives
Upon completion of the reading content, the healthcare professional will be able to:
Define absorption, distribution, metabolism, and excretion.
Identify and differentiate between pharmacokinetics and pharmacodynamics
Describe the dosage response curves
Identify the importance of half-life and clearance of a drug
Recall the P450 metabolic pathway
Policy Statement
This activity has been planned and implemented in accordance with the policies of FastCEForLess.com. If you want to review our policy, click here.
Disclosures
Fast CE For Less, Inc. and its authors have no disclosures. There is no commercial support.
To access these Fast Facts, purchase this course or a Full Access Pass.
If you already have an account, please sign in here.
To access these Fast Facts, purchase this course or a Full Access Pass.
If you already have an account, please sign in here.
| Affinity | The degree of drug receptor interaction for a drug and protein receptor population, where the tissue response reflects the quantity of drug receptor complexes that are intact at any given moment. |
| Agonist | A substance that binds to a specific receptor and triggers a response within the cell. It mimics the response of a hormone that binds to the same receptor, and in adequate concentrations can cause activation of all receptors; call a full agonist. |
| Agonist-antagonist | A substance or agonist that has receptor protein affinity but only 1/10th to 1/15th of the potency of a pure agonist. |
| Antagonist | A substance that has affinity for the receptor but does not have any efficacy in eliciting a response. It does not activate the receptor to produce a physiologic action, and by occupying the receptor, it blocks any agonists from binding to the receptor to produce an effect. Competitive antagonists are reversible, however noncompetitive antagonists are irreversible and require the synthesis of new receptors to reestablish homeostasis. |
| Chiral | A molecular system whose asymmetry results in handedness; the existence of a pair of nonsuperimposable mirror-image shapes, as illustrated by the relationship between one’s right and left hands. |
| Clearance | A pharmacokinetic measurement of the volume of plasma from which a substance is completely removed per unit time. |
| Competitive Antagonism | A drug or chemical that binds to a specific receptor site and once there, blocks another drug or chemical from binding to that site. |
| Context Sensitive Half-time | The time taken for blood plasma concentration of a drug to decline by one half after an infusion designed to maintain a steady state (a constant plasma concentration) has been stopped. The “context” is the duration of infusion. |
| Efficacy | A drugs ability to produce a desired effect through stimulation of a receptor. It describes the maximum effect that can be achieved with a drug. |
| Effective Dose | The quantity of a drug that will produce the effects for which it is administered. |
| Enantiomerism | The relationship between two stereoisomers having molecules that are mirror images of each other. Enantiomers have identical chemical and physical properties in an achiral environment but form different products when reacted with other chiral molecules. |
| Extraction Ratio | A measure in renal physiology that is used to calculate renal plasma flow to evaluate renal function. It measures the percentage of the compound entering the kidney that was excreted into the final urine. |
| First Pass Hepatic Effect | The rapid uptake and metabolism of an agent into inactive compounds by the liver, immediately after enteric absorption and before it reaches the systemic circulation. |
| Inverse Agonist | A drug that binds to a receptor and causes an opposite action of an agonist. |
| Isomer | One of two or more compounds, radicals, or ions that contain the same number of atoms of the same elements but differ in structural arrangement and properties. |
| Lethal Dose | Represents a dose, usually recorded as dose per kilogram of subject body weight, at which a given percentage of a population. |
| Ligand | A molecule that can bind and form a complex with a receptor to produce a biologic response. Ligands are neurotransmitters and hormones that are drugs. |
| Ligand-gated Ion Channel | A group of transmembrane ion-channel proteins that open to allow ions such as Na⁺, K⁺, Ca²⁺, and/or Cl⁻ to pass through the membrane in response to the binding of a chemical messenger (a ligand), such as a neurotransmitter. |
| Noncompetitive Antagonism | An antagonist that binds to a receptor and because of its occupancy, blocks an agonist’s activation of that receptor to the effect that no amount of agonist present in the receptor compartment can overcome this antagonism. |
| Partial Agonist | A partial agonist activates a receptor but cannot produce a maximum response; it has lower efficacy than a full agonist. It can also partially block the effects of a full agonist. |
| Pharmacodynamics | The branch of pharmacology concerned with the effects of drugs and the mechanism of their action. |
| Pharmacokinetics | The branch of pharmacology concerned with the movement of drugs within the body. |
| Potency | A term used to differentiate between different agonists that activate the same receptor and can produce the same response, but at different concentrations. The most potent drug requires the lowest does of medication to elicit a desired effect. |
| Protein Binding | Refers to the degree to which medications attach to proteins within the blood. A drug’s efficiency may be affected by the degree to which it binds. |
| Racemic Mixture | A mixture that has equal amounts of left and right-handed enantiomers of a chiral molecule. |
| Receptor | A chemical structure that is composed of a protein that receives and sends signals that may be integrated into biological systems. |
| Stereochemistry | Involves the study of the relative spatial arrangement of atoms that form the structure of molecules and their manipulation. |
| Stereoselectivity | The property of a chemical reaction in which a single reactant forms an unequal mixture of stereoisomers during a non-stereospecific creation of a new stereocenter or during a non-stereospecific transformation of a pre-existing one. |
| Tolerance | An occurrence that requires increasing concentrations of a drug to produce a desired response. Tolerance usually occurs because of chronic exposure to the agonist. |
Pharmacodynamics is the study of how drugs effect the body. ² The most common mechanism is through the interaction of a drug with tissue receptors that are either in cell membranes or in the intracellular fluid. The extent of receptor activation and the subsequent biological response is related to the concentration of the activating drug, namely the agonist. This relationship is described by the dose–response curve. ² The curve plots the drug dose (concentration) against its effect. This pharmacodynamic relationship can be influenced by factors such as age and disease, and by the presence of other drugs that compete to bind at the same receptor, also called the receptor antagonist. Some drugs acting at the same receptor differ in level of the biological responses that they can achieve (efficacy) and the amount of the drug required to achieve a response (potency). Drug receptors can be classified based on their selective response to various drugs. Constant exposure of receptors or body systems to drugs sometimes leads to a reduced response (desensitization). ¹
A receptor is a single protein that a drug can attach itself to. ² There are a variety of receptor types that have been isolated and investigated, including opioid, acetylcholine, histamine-subtypes, benzodiazepines, GABA, nicotinic, muscarinic, the pain related capsaicin receptor, and countless others. There are four types of regulatory proteins that are receptors: cell surface proteins, carrier molecules (also known as transporters), enzymes, and ion channels. ⁴ Drug receptor proteins are located within the luminal membrane and the surface of the ion channel. Having a complete saturation of the receptor with drug molecules is not necessary for a desired medication response. For instance, when a drug is administered intravenously, the amount needed for maximal tissue response is delivered to the receptor within the time needed for complete circulation; approximately 1 minute.
| Cell Surface Receptors | G-protein-coupled receptors: Are receptors for hormones (neurotransmitters, amino acids, biogenic amines), and neuropeptides -Activate/inhibit adenyl cyclase and guanylyl cyclase, which generate cyclic AMP and cyclic GMP, respectively -Activate phospholipase C -Modulate ion channels |
| Enzyme-Linked Cell Surface Receptor | Catalyze biochemical reactions, some of which involve the production of key mediators of physiological processes in body systems. A drug will interfere with the active site of the enzyme or affect co‐factors that is needed by the enzyme for activity. In most cases blocking the active site is competitive, although in some cases it may be long-lasting and effectively irreversible. Receptor-guanylyl cyclase: Are receptors for atrial natriuretic peptide Receptor serine/threonine kinases: Are receptors for activin, inhibin, Mullerian inhibiting substance, and transforming growth factor Receptor tyrosine kinases: Are receptors for peptide growth factors Receptor tyrosine phosphates: Are ligands that are mostly unknown Tyrosine kinase-associated: Are receptors for cytokines, growth hormone, and prolactin. |
| Intracellular Receptors | Binding of a ligand will create or block synthesis of a new protein, which may take hours or days to create a biological effect. Steroid receptor superfamily: Are receptors for steroids, sterols, retinoic acid, thyroxine, and Vitamin D. |
| Kinase-linked receptors | Linked directly to an intracellular protein kinase that triggers a cascade of phosphorylation reactions. |
| Ligand-gated ion channels: | Are receptors for neurotransmitters (amino acids, peptides, and biogenic amines) -Found in excitable tissues and a potential target for drugs that can block the channel or interfere with conductance in other ways -Mediate fast synaptic transmission |
| Transporter proteins | Specialized proteins that carry ions or molecules across cell membranes. Movement may be in either direction, and may involve exchange of one substance for another, co-transport of two or more substances in the same direction, or ‘pumping’ of a single substance into or out of a cell or organelle. Drugs may act on transporters to inhibit their activity or may also act as ‘false substrates’, preventing the transport of the normal biological substrate. |
Receptors are proteins that are bound, partially bound, or unbound by a ligand (drug). When the receptor is bound to the agonist ligand (a drug that activates the receptor), the effect of the drug is produced. ⁵ The magnitude of the drug’s effect is reflective of the total number of receptors that are bound. A receptor can also be bound by an antagonist ligand (a drug that binds to a receptor without activating it). An antagonist ligand can block the action of the agonist ligand by getting in the way of it and preventing the agonist ligand from binding to the receptor and producing a drug effect. Increasing concentrations of the antagonist ligand will progressively block the response to the agonist ligand on the receptor² (think of Narcan’s effect on narcotics). This antagonistic effect is called competitive antagonism. Noncompetitive antagonism occurs when after the administration of an antagonist ligand; high concentrations of an agonist ligand are ineffective in overcoming the antagonistic response due to irreversible binding, or binding to a different location on the receptor. Simply stated, non-competitive antagonists reduce the magnitude of the maximum response that can be attained by any amount of agonist. Likewise, a partial agonist is a drug that binds to a receptor and activates it, but not as much as a full agonist. Even in large doses, a partial agonist cannot create the full agonist effect. Partial agonists may also have antagonist activity, called agonist antagonist. When a partial agonist is administered with a full agonist, it decreases the effect of the full agonist. For example, butorphanol is a partial agonist on the opioid receptor. When given with fentanyl, butorphanol will partially reverse the effects of fentanyl, to the extent of causing withdrawal in a chronic opioid user.
The drug response equation is fundamental to understanding pharmacology. It comes from the law of mass action and exemplifies how a drug (D) combines to a receptor (R) to form a drug receptor complex (DRC) that results in a tissue response (TR). The equation is as follows: ²˒⁴
D+R↔(DRC)↔TR
This is a unique equation because the drug receptor complex represents a highly selective process, however the response at the tissue level varies from person to person which is reflective of each person’s genetic profile, receptor profile, and physiologic state. Although individual responses to a drug may vary, the basic drug response relationship depends on a common pharmacologic theory of drug action called occupancy theory. This theory describes the magnitude of a drug’s effect and how it is proportional to the number of receptors a drug occupies. ³ This theory is useful when identifying therapeutic drug dosages for individuals within a “normal” population, however, may have some variance when identifying patients within the neonatal, pediatric, geriatric, or obstetric populations, or those with cardiac or other chronic diseases. An individual’s age, sex, body habitus and surface area, genetic profile, pathologic state, and basal metabolic rate all influence the pharmacological response to the administration of a drug/medication.
Desensitization describes the occurrence of the diminished physiological response to a drug when it is administered continuously or repeatedly. The physiologic response may be increased by increasing the drug dosage or concentration, however in some cases, increasing the dosage or concentration will have no effect. ¹˒³ Tachyphylaxis describes desensitization that occurs rapidly, often after the first dose of a drug is administered. Tolerance is most used to describe a gradual loss of physiological response to a drug that occurs over a long period. Drug resistance describes a loss of effectiveness of a drug. Desensitization may occur because of established chemical, hormonal, and physical changes that offsets a drug’s action. However, the cause, abruptly discontinuing the drug can cause a rebound effect of withdrawal.
The administration of a drug is mainly determined by a mean therapeutic dose per kilogram of body weight or body surface area that is calculated from a previously determined dose for the average population. Unfortunately, this method may be responsible for the overdosing or underdosing of patients because of variations in the population that are not considered. To prevent the occurrence of overdosing or underdosing when administering intravenous drugs/medications, the licensed healthcare provider must titrate the drug administration until a therapeutic response is reached. There are two types of dose-response curves; graded and quartal, that describe the average drug response with and variability within a population.
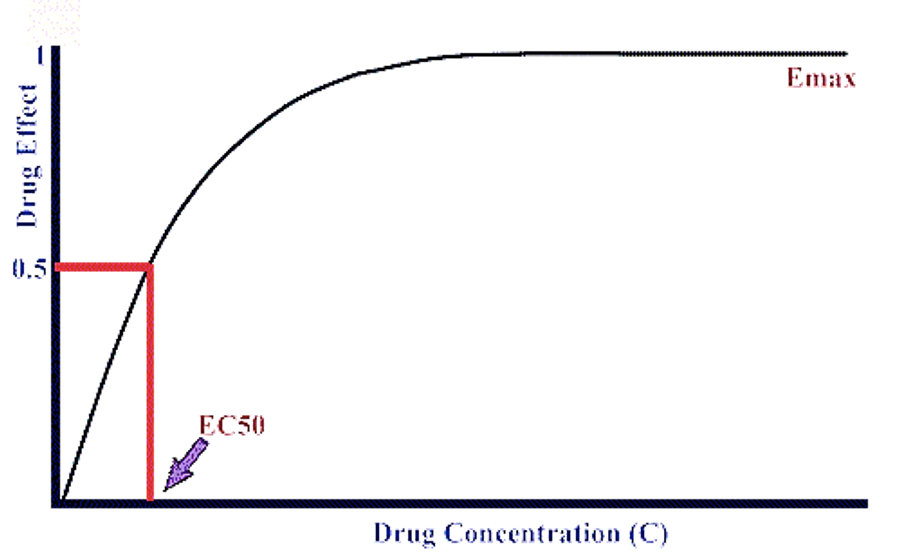
The graded dose response curve characterizes the changes of a drug’s concentration in a measured response when a dose of intravenous drug is increased.² The curve has a hyperbolic shape and exemplifies the greatest change in drug concentration response that occurs to the left on a small portion of the x-axis. A plateau in the line will occur when the highest dose is administered, and the highest response is achieved. Usually, when 20-80% of the maximal response to a drug is reached, the curve will turn into a straight line as a result of a proportional relationship between the changes in the dose of the drug administered, and the response that the dose elicits.
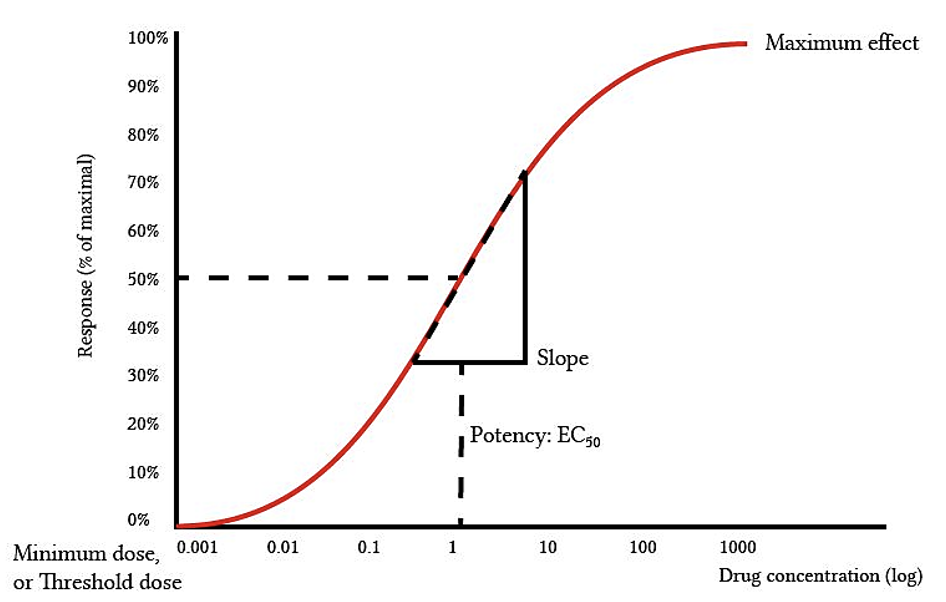
The quantal dose response curve provides information on the frequency with which an administered drug dose produces a desired response to similar individuals within a population and describes how there are variations in responses to a drug when administered at a threshold dose.² For example, when administering propofol for induction of anesthesia, a small number of people will become unconscious 0.5mg/kg is administered, more people will become unconscious when 1mg/kg is administered, and virtually everyone will become unconscious after the administration of 2 mg/kg of propofol. The variations in threshold dosages to gain the required response (unconsciousness) produces an S-shaped curve. Information regarding the population’s dose response characteristics can be gained from quantal dose response curves. For instance, the effective dose, toxic dose, and lethal dose can all be identified. The effective dose 99% (ED₉₉) and lethal dose 1% (LD₁) are both used to identify the therapeutic safely margin of a drug and is shown in the following equation²:
Safety Margin = LD₁ − ED₉₉/ ED₉₉ x 100
When the therapeutic safety margin of a drug is high, the risk of drug induced death is small. The opposite is also true; when a therapeutic safety margin is low, the risk of drug induced death is high.
Another descriptor that can be obtained from the quantal dose curve is the median effective dose (ED₅₀), which is the dose at which 50% of the population will have the desired effect.¹˒² The ED₅₀ is most often used to compare the potency of drugs within a class. Comparisons of drugs within a class can be made because the ED₅₀ dose is derived from the linear portion of the curve that represents 20-80% of the maximal response to a drug. It should be noted that this represents the median or average response to a drug within a population of similar individuals, however everyone within a population responds to a drug as a result of his or her biologic and genetic variations in drug receptor proteins.
The 50 percenters²:
• Context sensitive half-time is the time for the plasma concentration of a drug to decrease by 50% from an infusion. The ‘context’ is the infusion and the half-time increases as the duration of the infusion increases because it takes longer for the concentrations of a drug to decrease due to accumulation in peripheral tissues.
• The effective dose (ED₅₀) is the dose of a drug that is needed to produce a desired effect in 50% of people receiving the drug.
• The lethal dose (LD₅₀) is the dose of a drug that would produce death in 50% of people receiving the drug.
• The therapeutic index is the ratio between the effective dose and lethal dose (ED₅₀/LD₅₀). The larger the therapeutic index is, the safer the drug is for administration.
A desired tissue response is achieved when enough free receptors are occupied and activated by a free drug. This idea holds true that at steady state, equilibrium exists between bound and unbound drug receptors and the concentration of free unbound drug at the receptor site. Key terms for receptor interactions include affinity, efficacy, drug receptor response triad, pure antagonist, agonist-antagonist, physiologic antagonism, and chemical antagonism. Please review the key terms at the beginning of chapter for a full list of definitions.
Pharmacokinetics describes the study of the changes in the concentration of an administered drug that occurs during absorption, distribution, metabolism, and elimination. ² This description is important, because the pharmacokinetics of a drug influences onset, duration of action, and offset of a drug, regardless of the route of drug administration. Once the drug is in the blood, it can either remain within the vascular system and bond to proteins within a body of water, or cross membranes and enter tissues. The unbound drug enters muscle, fat, organs, and receptors. The transfer of the drug depends on may factors that include the molecular size of the drug, lipid solubility, and protein binding. Uptake of the drug is also influenced through blood flow to the tissues and the concentration gradient of the drug across cellular membranes.
A drug is better able to cross the lipid barrier and tissue membranes when it is small. When a drug is administered, it will be absorbed across a biologic membrane with small pores or openings. ³ A molecular weight greater than 200 generally is incapable of crossings membrane. Molecules can be transported across a membrane via active or passive transport. Passive transport does not require energy and allows transfer of a drug from an area of high concentration to an area of lower concentration, whereas active transport is the fastest and requires energy for transport. The active transport system utilizes carriers that form complexes with the drug molecules that are located on the membrane surface and involve movement of the drug molecule against a concentration gradient from an area of low concentration to an area of high concentration.
Most drugs are either weak acids or weak bases, and when administered they behave as a chemical in a solution in ionized and nonionized forms. ²˒⁴ The ionized form (charged form) is water soluble, and the nonionized form (uncharged form) is lipophilic. The lipophilic molecules are lipid soluble, and thus can diffuse across cellular membranes (blood-brain, gastric and placental barrier). Opposite to this, an ionized drug is water soluble and unable easily cross the cell membrane. The higher the degree of ionization, the less access the drug has to the cell membrane, which is important, because ionized drug are not absorbed well when taken by mouth and may not be metabolized by the liver, instead being excreted by the kidneys.
Changes in protein binding can influence a drugs clinical effect. The number of binding sites on a plasma protein for a drug is infinite. ⁵ Binding can be overcome by adding more drugs. When the plasma concentration of a drug is reduced, or when a second drug is administered that binds with the same plasma protein as the first drug, the first drug can dissociate itself from the plasma protein. For example, if a drug is used chronically and is at steady state, an equilibrium will be reached. If a new drug with a high affinity for the same protein is administered, the new drug will compete with the old drug for binding sites. This will cause a displacement of the first drug and an increase in free fraction of the first drug. Protein binding is expressed as the percentage of the total drug that is bound. Drugs that have protein binding that is greater than 90% are noted to have an increase in effect if they are displaced from plasma proteins. Opposite to this, drugs that have less than 90% protein binding have little to no change in effect when displaced from plasma proteins.
The route that a drug is administered will determine how much of the drug will be delivered to systemic circulation. When the full amount of a drug that is administer is delivered to systemic circulation, it is described as having 100% bioavailability. ²This most often occurs with intravenous administration of a drug. Drugs can be administered via oral, sublingual, injection, transdermal, rectal, intramuscular, subcutaneous, or inhalation route. It should be noted that there is no absorption of a drug if it is administered intravenously.
- The oral route (enteral) is the most common and convenient route for administering drugs. Oral drugs, however, tend to have a lower bioavailability because of changes in the gastrointestinal environment because of physical activity and food intake. Drug absorption can also be varied due to the large surface area of the stomach and the length of time that a drug remains there. The stomach has a low PH (1.5-2.5) and drugs that are acidic are likely to be highly absorbed. Base drugs are less likely to be absorbed in the stomach, and more likely to pass through the stomach and be absorbed in the intestines because of the alkaline environment and vascularity.
- First pass hepatic effect occurs when a drug that is absorbed in the gastrointestinal tract enters the portal venous blood and passes through the liver for metabolism before entering systemic circulation and being delivered to tissue receptors. Oral dosages must be increased to overcome this phenomenon.
- Sublingual and buccal routes of drug administration bypass the first pass hepatic effect and are delivered to the superior vena cava for transport to the desired site for effect.
- Drugs are often administered rectally to prevent emesis that could result from irritation of the gastrointestinal mucosa of a drug, or when oral ingestion of a drug is not feasible.
- Injection (parental) administration of a drug is the most reliable route of administration with the fastest onset.
- Subcutaneous administration occurs when the drug is placed under the skin.
- Intramuscular administration occurs when a drug is placed deep into the muscle among the muscle fascicles.
- Drugs that are administered subcutaneous and intramuscular routes rely on capillary blood flow and lipid solubility to reach systemic circulation.
- Topical (transdermal) administration of a drug is usually occurs when sustained release is needed for steady therapeutic plasma concentrations.
- Inhalation (pulmonary) administration of a drug propels aerosols into the alveolar sacs. The lungs have a large surface area for drugs to be absorbed.
Bioavailability describes how much of a drug reaches the effect site after it enters the circulatory system. ⁵ A drugs rate of systemic absorption will determine the intensity and duration of action that it exhibits. Various drug routes and approximate bioavailability are as follows:
| Route | Bioavailability% |
| Intravenous | 100 |
| Topical | 80-100 |
| Intramuscular | 75-100 |
| Subcutaneous | 75-100 |
| Sublingual | 60-100 |
| Rectal | 30-100 |
| Inhalation | 5-100 |
| Oral | 5-100 |
Compartment models exhibit the body as distinct sections with calculated volumes. Compartment models are useful in predicting changes in drug concentrations and serum concentrations in tissues. ²
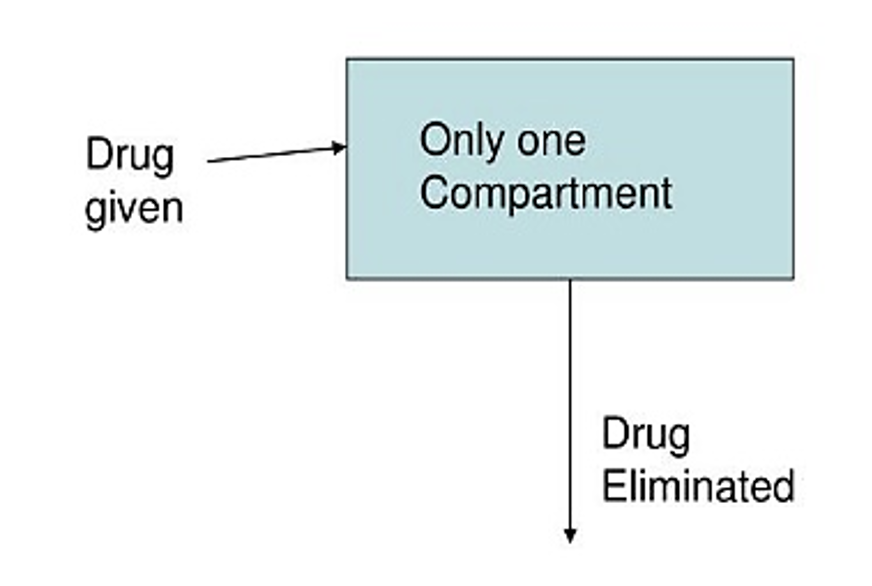
The entire body is representative of the single-compartment model. When a drug is administered it is assumed that it is distributed to all areas of the body. There are some instances when a drug is not immediately administered throughout the body, and in this instance, a two-compartment model is needed.
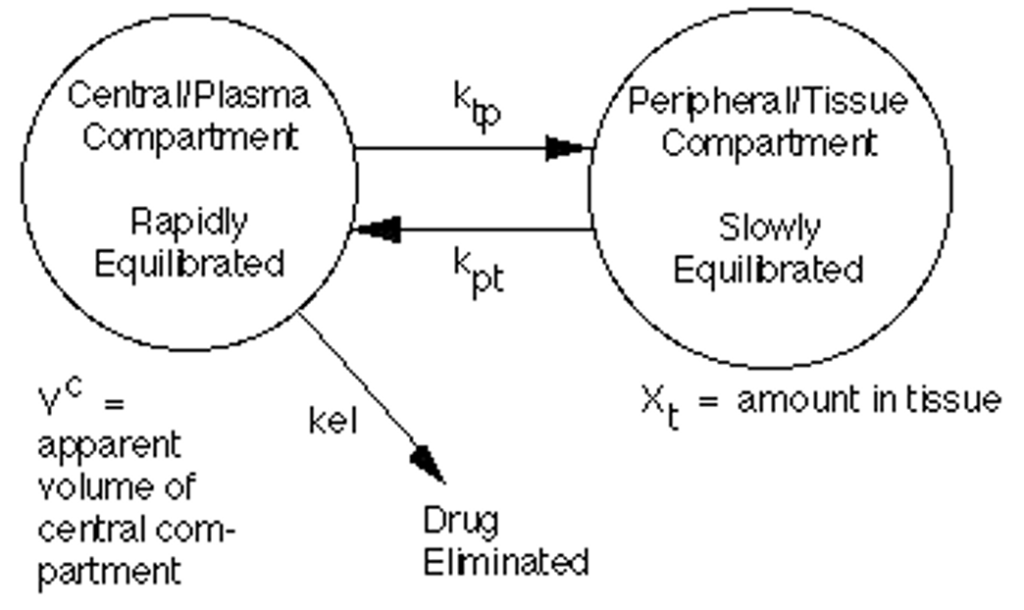
In a two-compartment model, the first compartment (central compartment, also known as the vessel rich group) is made up of intravascular fluid, the heart, lungs, brain, liver, and kidneys. The central compartment only represents 10% of body mass, but it receives 75% of the total cardiac output. The peripheral compartment (vessel poor group) is made up of fat, muscle, and bone. The peripheral compartment represents 90% of the body mass but only receives 25% of the total cardiac output.
When an intravenous drug is administered, it initially enters the central compartment were organs with the highest blood flow are located. The highly perfused organs equilibrate with the initial high serum concentrations of a drug. ³ As blood flows through the peripheral compartment, the concentration of the drug rises more slowly and does not reach the same concentration as the central compartment. As blood continues to flow through the tissues of the peripheral compartment, plasma concentration of th drug decreases, which is called the alpha half-life of a drug. This cycle will continue; the drug will reemerge from the central compartment to the peripheral compartment until the drug is eliminated. This allows for the calculation of the volume of distribution.
The volume of distribution describes the volume of a drug that is distributed after it has been administered. It is used to calculate the loading dose of a drug that will achieve a steady state concentration. The equation is as follows²:
Volume of Distribution = Dose of a drug/ Plasma concentration of a drug
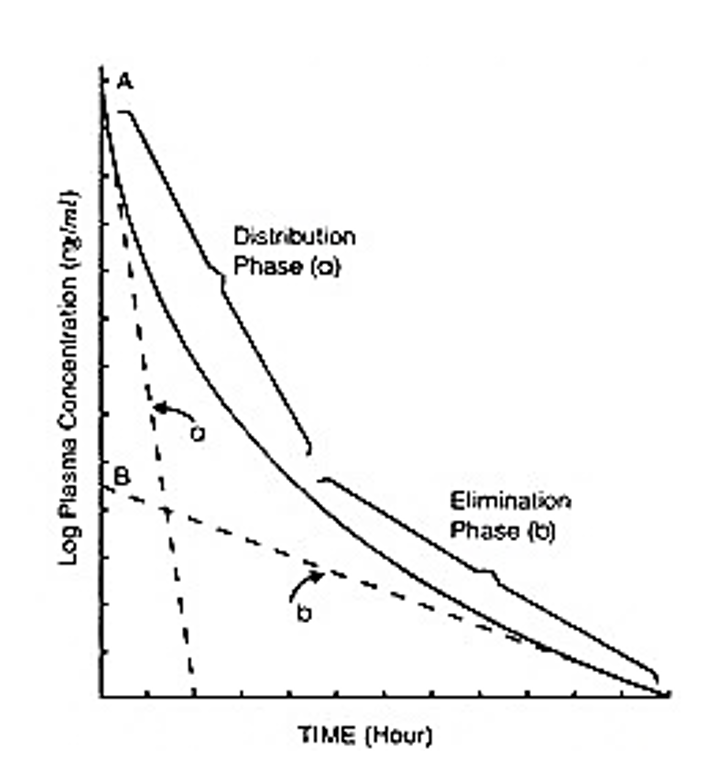
The plasma concentration curve represents the decline of the plasma concentration of a drug after rapid intravenous administration. ² The y axis of the graph represents the plasma concentration. The x axis of the graph represents the amount of time after an intravenous drug is administered. The a axis; the distribution phase, represents the initial transfer of the drug from the central compartments to the peripheral compartments. The line has a sharp slope that represents lipid soluble drugs that can easily transfer across cell membranes and be distributed to peripheral compartments rapidly. The b axis is a slower slope that represents elimination. Once equilibrium of a drug occurs, the concentration of a drug will continue to decline because of elimination from the liver, kidney, and other organs. The elimination phase of the plasma concentration determines the elimination half-life of a drug, which becomes important when determining dosage intervals of a drug
Steady state occurs when a stable plasma concentration of a drug occurs. ¹ This results from the equilibration that occurs from continuous drug administration. Concentrations of a drug may vary between organs, but the concentrations within each of the organs does not change. This is a result of the amount of a drug being administered being equal to the amount of drug being eliminated. Steady state occurs with continuous intravenous administration of a drug.
Metabolism is the enzyme-catalyzed structural change of a drug that usually occurs with more than one pathway.² The main organ for metabolism of a drug is the liver, however metabolism can also occur in the kidneys, gastrointestinal tract, plasma, heart, brain, lungs, and skin. The goal of metabolism is to change lipid soluble drugs into water-soluble forms so that they may be eliminated from the body by the kidneys. When administered in a therapeutic dose, a drug will be metabolized through first order process; an occurrence where the drug is cleared at a rate that is proportional to the amount of a drug that is available in the plasma. For instance, a dug with a high plasma concentration will be metabolized at a faster rate. Drug metabolism occurs in two phases: Phase I reactions and Phase II reactions. ²
- . The result of phase I reactions is a polar compound that is easily excreted by, the kidneys.
- The molecule of oxygen then splits, causing one atom to oxidize each molecule of a drug and the other atom to incorporate into a molecule of water. The loss of electrons will result in oxidation.
- The new compound is the new form of the drug or the drug product of the Phase I reaction and has little to no biologic activity. A conjugation reaction results in a more polar compound that is more easily extractable by the kidney via glomerular filtration.
- Hepatic microsomal enzymes are mainly located in the smooth hepatic endoplasmic reticulum. These enzymes are called microsomal enzymes because they are fragments of the endoplasmic reticulum that are obtained in vitro. The microsomal fraction includes proteins called cytochrome P-450.
- Hepatic enzyme availability and activity varies from person to person and is determined genetically, however enzyme activity can be stimulated over a period with continued exposure to a drug, resulting in enzyme induction. For example, a chronic alcohol user will have increased enzyme activity, which will result in increased drug metabolism.
- Hepatic microsomal enzymes are mainly located in the smooth hepatic endoplasmic reticulum. These enzymes are called microsomal enzymes because they are fragments of the endoplasmic reticulum that are obtained in vitro. The microsomal fraction includes proteins called cytochrome P-450.
Elimination half-life is the time needed for the plasm concentration of a drug to decrease by half after a rapid bolus administration. ¹A drug is considered as being fully eliminated when approximately 95% of it has been eliminated from the body. This usually occurs within five half-lives. This is important, because administering intravenous drugs more frequently than elimination will lead to drug accumulation and potential overdose. Other elimination models include:
- Zero-order elimination: A constant amount of a drug is eliminated over time, and it is not a percentage.
- Michaelis -Menton model: dose dependent and follow zero-order elimination at high doses and follow first order metabolism once drug levels have fallen.
The clearance of a drug is controlled by the properties of a drug and the body’s ability to eliminate it.³˒⁵ Clearance is defined as the volume of plasma that is cleared of a drug by metabolism and excretion per unit of time (Clearance=QxE). Clearance is an important concept because it influences the steady state concentration of a drug that is administered by infusion or at repeat intervals. The two main organs for clearance are the liver and kidneys.
- Hepatic clearance: Drugs that have a high extraction ratio of 0.7 or greater are require liver perfusion to be cleared. Hepatic blood flow exceeds enzyme activity in clearing drugs from the body. A decrease in blood flow will decrease the rate of clearance, and opposite to this, a high lever perfusion rate will lead to faster drug clearance. This is termed perfusion-dependent elimination.
- Capacity elimination can occur in drugs that have an extraction ratio of 0.3 or less. When a drug has a low extraction ratio only a small portion of it will be removed per unit of time. Changes in hepatic perfusion will not have a significant effect on hepatic clearance. Instead, hepatic clearance is dependent on hepatic enzymes and the degree of protein binding. As a result, an increase in enzyme activity can cause faster elimination of a drug from the body, and enzyme suppression can cause slower elimination.
- Renal clearance occurs when the kidneys excrete water-soluble drug molecules. The excretion of the molecules involves glomerular filtration, active tubular secretion, and reabsorption. Water soluble drugs are filtered by the glomeruli and eliminated. The kidneys do not excrete lipid-soluble drugs efficiently, and as a result they are reabsorbed from the renal tubules back into systemic circulation to be metabolized by the liver into water-soluble molecules.
Knowledge and competency in pharmacodynamics and pharmacokinetics are essential when administering drugs. The absorption, distribution, metabolism, and excretion of a drug is influenced by many factors, such as a drug’s lipid solubility, molecular size, surface area for absorption, hepatic and renal function, etc. There are many ways that can predict the effect and metabolism rate of a drug, and these ways are important in preventing overdosing of medications. To prevent overdose and improve safe practices when administering drugs, the healthcare provider must consider the effects of the drug administered and how it is metabolized and eliminated.
- Duffus, J. H. (2016). Dose-response curve. IUPAC Standards Online. doi:10.1515/iupac.65.0417
- Nagelhout, J. J., Plaus, K., & Kaiser Permanente School of Anesthesia Sass Elisha. (2017). Nurse anesthesia. Philadelphia, MO: Saunders.
- Pharmacodynamics. (2020). doi:10.4135/9781529728545
- Willihnganz, M., Gurevitz, S. L., & Clayton, B. D. (2019). Clayton’s basic pharmacology for nurses. Maryland Heights, MO: Mosby.
- Yeager, J. J., Burchum, J., & Rosenthal, L. (2015). Study guide for pharmacology for nursing care. St. Louis, MO: Elsevier Health Sciences.
To access these Fast Facts, purchase this course or a Full Access Pass.
If you already have an account, please sign in here.